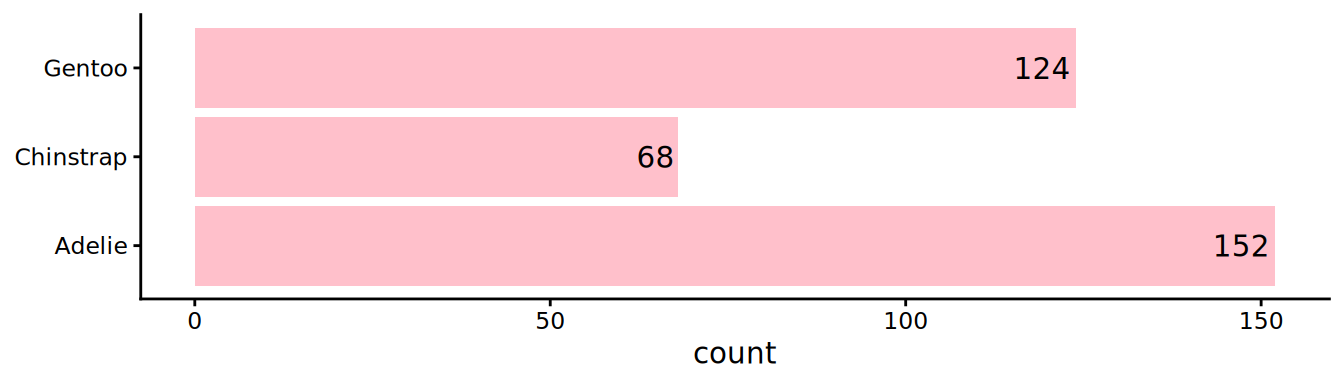
# y mapping to get flipped axis
ggplot(penguins, aes(y = species)) +
# geom_bar does count how many penguins are observed for each species
geom_bar(fill = "pink") +
# add count in bars, label is counting, and horizontal adjust is 1.1 to be just before bar height
geom_text(aes(label = scales::comma(after_stat(count))), hjust = 1.1, stat = "count") +
# light theme
theme_classic() +
labs(y = NULL)